为了查找某个研究领域的相关信息,生物学家往往要花费大量的时间。与此同时由于不同数据库之间的信息可能不同步或者术语不一样,这使得信息的检索更加的麻烦。如果让人来做还勉强能查,但是让机器来查询的话一切就会变的无章可循。Gene Ontology(GO)就是为了解决这种问题而发起的一个项目;每一个GO Term都是由7个数字标识这个Term编号,同时还有一个lable标识具体的生物学功能。每个Tern属于一个本体,在GO中有三个ontology它们分别是:
- molecular function,
- cellular component
- biological process
今天要做的就是大批量的根据某个功能的关键字,搜索已经报导的基因对应的序列信息;从而进行Blast找到你所研究的物种中的同源基因。这里以爬取Ethylene关键字为例,分以下三个步骤搞:
- 根据关键字爬取GO编号信息
- 根据GO编号爬取对应的基因注释信息
- 根据基因注释信息爬取基因的序列信息
1.爬取GO编号信息
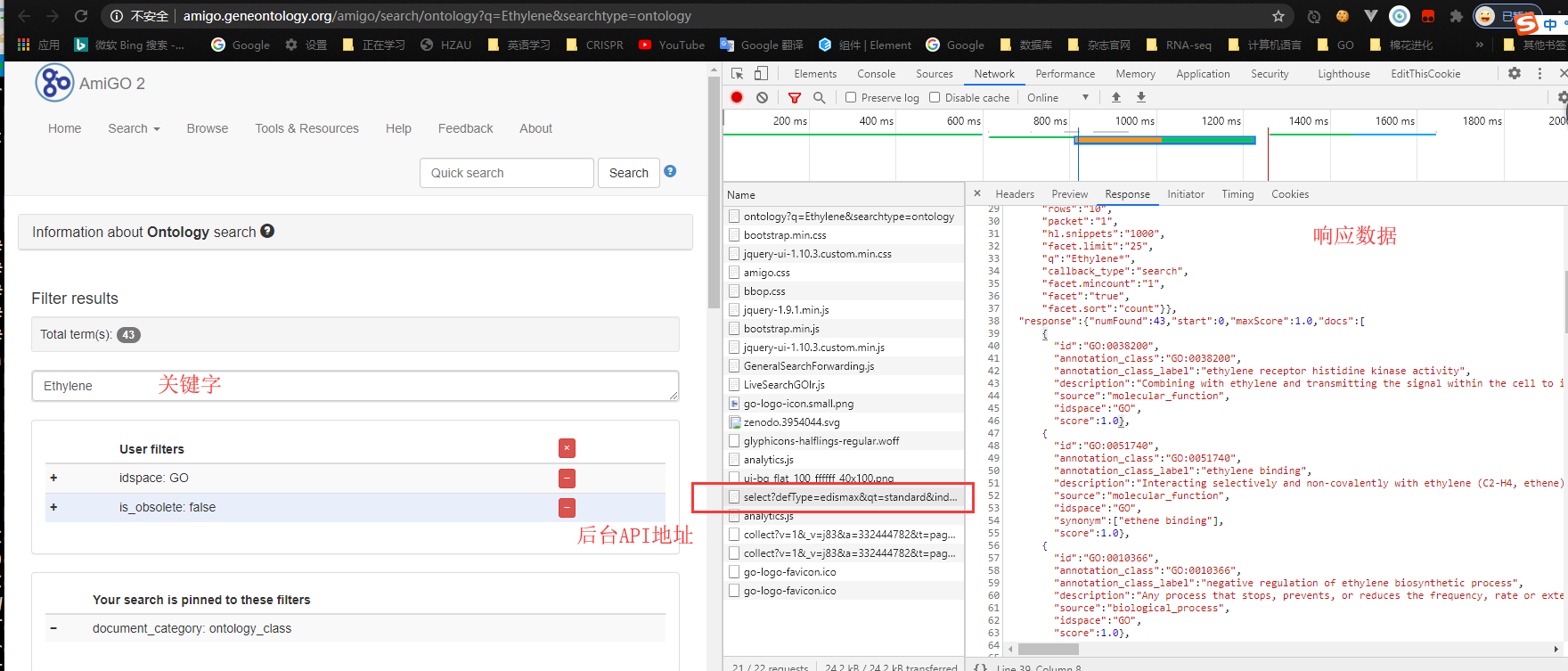
网页地址: http://geneontology.org/
![af0Pr4.png]()
在开发者模式下可用发现浏览器请求的API地址为
http://golr.geneontology.org/solr/select?defType=edismax&qt=standard&indent=on&wt=json&rows=10&start=0&fl=annotation_class,description,source,idspace,synonym,alternate_id,annotation_class_label,score,id&facet=true&facet.mincount=1&facet.sort=count&json.nl=arrarr&facet.limit=25&hl=true&hl.simple.pre=%3Cem%20class=%22hilite%22%3E&hl.snippets=1000&fq=document_category:%22ontology_class%22&fq=idspace:%22GO%22&fq=is_obsolete:%22false%22&facet.field=source&facet.field=idspace&facet.field=subset&facet.field=is_obsolete&q=Ethylene*&qf=annotation_class%5E3&qf=annotation_class_label_searchable%5E5.5&qf=description_searchable%5E1&qf=synonym_searchable%5E1&qf=alternate_id%5E1&packet=1&callback_type=search&json.wrf=jQuery214044957252147235494_1596787199447&_=1596787199448
![afwr8K.png]()
接下来使用python中的urllib包向目标API地址发起GET请求,并且使用Json包将响应的数据解析成Dict对象;
这里对同一个URL进行了两次请求,主要是为了获取所有的响应数据;因为每次请求时,默认只响应10条数据
1
2
3
4
5
| response = urllib.request.urlopen(getGoItemUrl)
jsonData = json.loads(response.read().decode('utf-8'))
return jsonData['response']['docs']
|
2.爬取对应基因编号
有的GO term可能没有对应的基因注释信息,有的GO term可能存在注释信息不完整的情况,统一使用NA进行填充
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
| for item in GoItem:
print("爬取GO编号:\t"+item['id'])
info = getGeneIdByGoItem(item['id'])
GoInfo = "\t".join([str(item[i]) for i in GoKeys])
if(len(info) == 0):
geneMessage = "\t".join(
['NA', 'NA', 'NA', 'NA', 'NA', 'NA', 'NA', 'NA', 'NA'])
out.append(GoInfo+"\t"+geneMessage+"\n")
else:
for geneItem in info:
geneMessage = []
for key in geneInfoKeys:
try:
geneMessage.append(geneItem[key])
except KeyError:
geneMessage.append("NA")
|
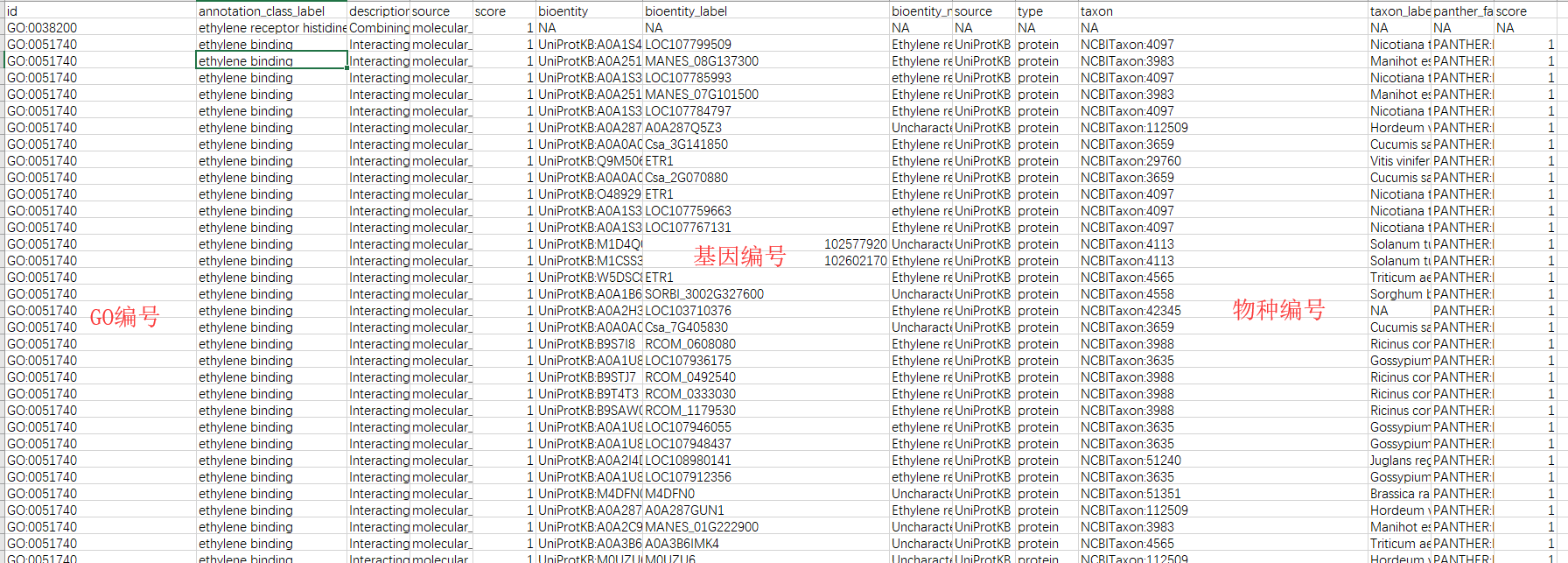
最终生成一个Txt文档;文档内包含GO Term的注释信息和对应的基因的注释信息
![afyF4s.png]()
3.爬取基因序列信息
有了上一步的GO Term的注释信息后,我想得到植物中所有与乙烯相关的基因的序列信息用于Blast;来获得我所研究的物种中对应的同源基因的功能。
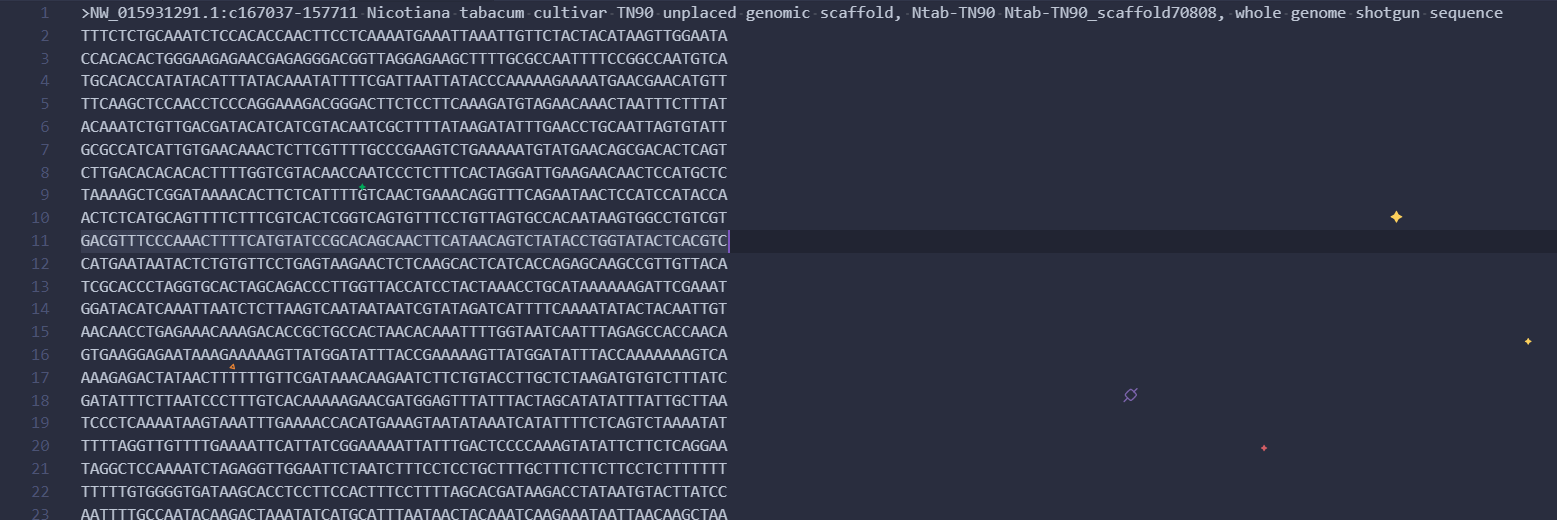
使用得到的基因编号和物种编号在NCBI中获得对应的gene序列信息,这里获得的序列都是正链上的序列,而不是有义链上的。
最终得到一个fasta序列文件
![af6TFH.png]()
4.脚本运行
当想要获取某一些感兴趣的基因序列信息时,只需要从第一个脚本生成的文件GOAnnotion.txt,截取对应的行到另一个文件,用作第二个脚本的输入文件
1
2
3
4
5
|
python GoItem2gene.py 关键字 GOAnnotion.txt
head 100 GOAnnotion.txt >interest_Annotion.txt
python getSeqByID.py interest_Annotion.txt interest_Annotion.fasta
|
![afRah4.png]()
脚本地址:https://github.com/BiocottonHub/zpliuCode/tree/8666815dfe1f14698596f160e236d9a6c5a042b7/GOTerm
GoItem2gene.py: 根据关键字获取基因编号
getSeqByID.py:根据基因编号提取基因序列
参考
- 基因本体概念