HiC-Pro安装与使用
HiC-Pro可以用来处理Hi-C数据,从原始的fastq文件(配对端Illumina数据)到标准化的交互图谱。简单的来说就是将Hi-C数据比对到拼装好的参考基因组上,并形成交互文件去存储Hi-C数据。下面我们就来介绍一下HiC-Pro的安装与使用。
HiC-Pro的安装
HiC-Pro所需要的这些依赖

1.使用conda安装依赖
python版本为2.7
1 | conda create -n hicpro python=2.7 #强烈推荐安装一个新的环境 |
2.安装对应的R包
在当前conda环境下运行R
也可以绝对路径运行R
1 | R |
3.配置confir-install.txt安装文件
PREFIX软件安装路径,会在该路径创建一个HiC-Pro_2.11.1目录
R_PATH指定conda环境下的R
PYTHON_PATH指定conda环境下的python
CLUSTER_SYSCLUSTER_SYS集群调度系统为TORQUE,SGE,SLURM,LSF四个中的一种
1 | PREFIX = /public/home/yxlong/yxlong/app/ #HiC-Pro所在目录 |
这些路径可以which+对应的依赖(如: which bowtie2)获得
4.安装
配置文件修改完成之后运行下列指令即可完成安装
1 | make configure |
HiC-Pro的使用
1.文件的处理
1.1文件夹的创建
建议创建两个文件夹命名为sample.reads和sample.ref(sample为自己的物种名字即可,下面以亚洲棉Ga为例)
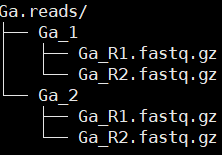
1.2Ga.reads的的目录结构
目录结构如下(依据自己的研究修改物种名即可,Ga_1、Ga_2…为不同的生物学重复)
1.3Ga.ref的相关文件
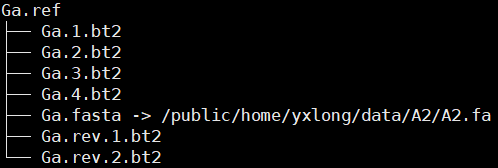
Ga.fasta文件是一个link文件,链接的是拼装好的参考基因组
建立索引 (索引的命名为物种名)
1 | /PATH/TO/bowtie2-build --threads 20 /Ga.ref/Ga.fasta /Ga.ref/Ga |
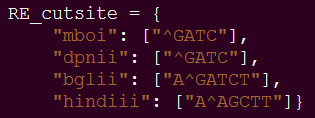
1.4酶切信息文件的获得
采用HiC-Pro自带的digest_genome.py程序酶切位点信息文件
1 | /PATH/TO/HiC-Pro_2.11.1/bin/utils/digest_genome.py -r hindiii -o Ga.HindIII.txt /Ga.ref/Ga.fasta |
1.5基因组中序列大小文件
序列大小文件格式如下 contig1[TAB]contig1_length
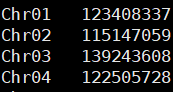
可以用如下指令获得
1 | samtools faidx /Ga.ref/Ga.fasta && awk '{print $1"\t"$2}' /PATH/TO/Ga.fasta.fai > Ga.fasta.size #注意生成的.fai在.fasta文件所在的目录下面 |
至此,相关文件的准备就结束了。
2.配置文件的书写
首先将配置文件复制一份到当前目录下
1 | cp /PATH/TO/HiC-Pro_2.11.1/config-hicpro.txt . |
然后,进行修改(以下为通常需要修改的参数,其他的参数的修改请参考官网)
1 | # N_CPU,例如N_CPU = 24 |
3.运行HiC-Pro
HiC-Pro的使用很简单,主要就是三个参数,指令如下(单线程运行)
1 | /PATH/TO/HiC-Pro_2.11.1/bin/HiC-Pro --input Ga.reads --output hicpro_output --conf config-hicpro.txt |
如果想多线程运行,要加上-p参数,输入指令后会在输出文件中生成两个文件HiCPro_step1.sh、HiCPro_step2.sh。先运行step1,step1执行完成后,再执行step2即可。

