Abstract
区分玉米不同单倍型的遗传变异对研究等位基因的功能和群体遗传分析,基因组的解析有着重要的作用。使用全长cDNA测序,在isform水平对两个玉米自交系品种和对应的杂交品种进行分析。通过使用IsoPhase方法,在胚胎、胚乳和根组织中验证了大多数从二代数据called SNP;并且在自交系与杂交后代之间鉴定了在等位基因间特异的、基因水平、isform水平的差异表达。在互惠杂交后代中,经过分型后对6907个基因注释了SNP,鉴定了大效应基因。并且,在不同的组织中鉴定了亲本起源的isform,杂交后代中独特的isform,印记基因。最后,作者表征了顺式和反式调节作用的变化。
introduction
对遗传变异进行分型,对于鉴定出潜在的致病变异,表征遗传变异和表型之间的关系有着重要的作用。玉米作为一个二倍体物种,虽然不同基因组之间存在很高的相似性,但是仍旧表现出等位基因的特异性表达ASE(等位基因间表达水平不相同)。并且ASE随着不同组织、不同发育阶段、不同生长环境中变化。等位基因在序列水平上产生不同的转录本,为了更好的解析玉米的转录图谱,因此有必要分析ASE。之前的研究是通过二代数据,在SNP水平对等位基因的表达进行量化,但是不能够提供全长的单倍型信息。
PacBio和Nenopore则通过对全长转录本进行测序,广泛的被应用于基因组注释中;但是只有少量的研究使用long reads进行isform水平的亚型研究。本研究中使用PacBio和RNA-seq测序,并且开发了一套流程IsoPhase,对杂交种中等位基因的亚型进行分型。第一个在玉米中实验IsoPhase方法进行研究,这套流程同样适用于多倍体物种。这套流程不需要父母本的信息,虽然父母本的信息可以区分母本还是父本的等位基因。并且isform水平可以确定到底是那个等位基因在转录,可以产生特定的等位基因转录组。
Result
全长转录组测序和生信分析流程
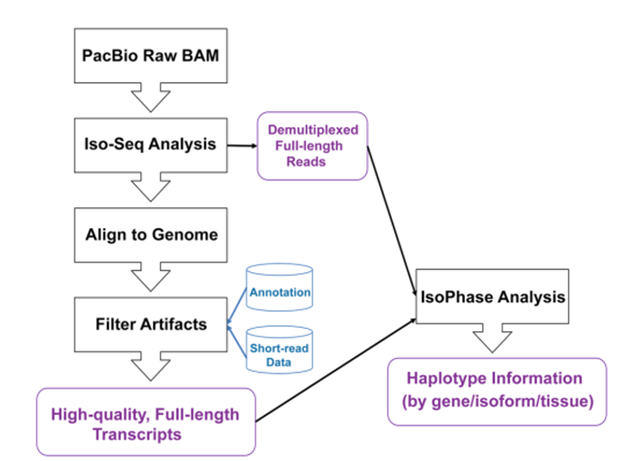
首先两个亲本在一些性状上存在巨大的差异,使得这两个亲本的杂交非常的适合研究等位基因的杂种优势。同时为了量化不同组织中基因表达谱,作者提取了不同物种、不同组织的RNA进行反转录,加上组织特异的barcode进行混池测序,将得到的环形共识序列使用Isoseq3分析流程。其中有76.3的read被归类为全长的转录本,根据barcode和ployA是否出现。经过Isoseq处理后,产生了25万个高质量的全长转录本。将这些全长转录本比对到玉米参考基因组上,约有99.3%被比对到参考基因组上,根据覆盖度和相似度进行筛选后,仅仅保留91.8%,这些序列进一步 collapsed 成9万多个非冗余的转录本。将没有比对上的全长转录本比对到 RefSeq NR database 数据库中其中约有50%的比对到了其他生物中,说明这些在文库制备的时候来至于质粒或者其他来源。
Isoform characterization
使用 SQANTI 将转录本与参考基因组注释进行比较,其中有 3344 被认为是新的没有被注释的转录本。其中有95.5%是位于基因间区的。
SQANTI 将转录本进行了分类
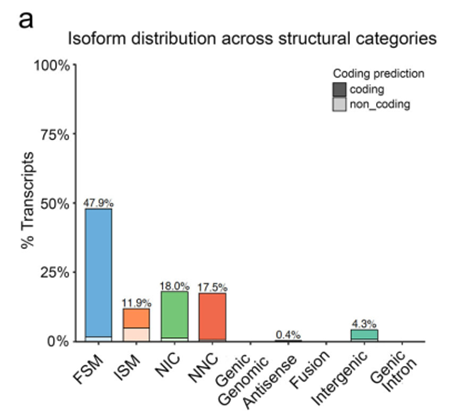
- FSM 与参考基因组注释的剪切完全匹配
- ISM 不完全匹配
- NIC 全新的在分类内
- NNC全新的不在分类内
- 反义链
- 基因间区
并且全长转录本测序的转录本长度能达到10kb以上。并且新鉴定出的基因主要是多外显子。
- 对于剪切位点完全匹配的isform,发生了起始和终止位点的改变。
将转录本进行分池,也就是分组织的分析。当一个组织中的转录本有一条全长转录本的read支持,就认为它已经表达了。每个组织中包含有20000到30000个表达的转录本。为了验证数据的饱和,对样本进行二次采样,Isoseq数据在基因水平上已经达到饱和。
在基因和isform水平比较双亲和杂交种,没有发现明显的模式。与此同时在比较双亲与两个杂交种时,发现一些共享的和品种特异的基因、isform;这个发现同样被二代测序的数据所确认。对这四个品种的剪切模式进行探讨发现,内含子保守时最主要的模式;使用二代数据进行定量分析发现,在三个组织中大多数基因表现出一种加性效应。同时比较了两个杂交种,发现它们在加性基因的数目上存在差异。这也可能是导致两个杂交种间存在差异的原因。在加性基因与非加性基因的比较中isform的数目上没有存在明显的差异。
全长转录本实现准确的亚型分型
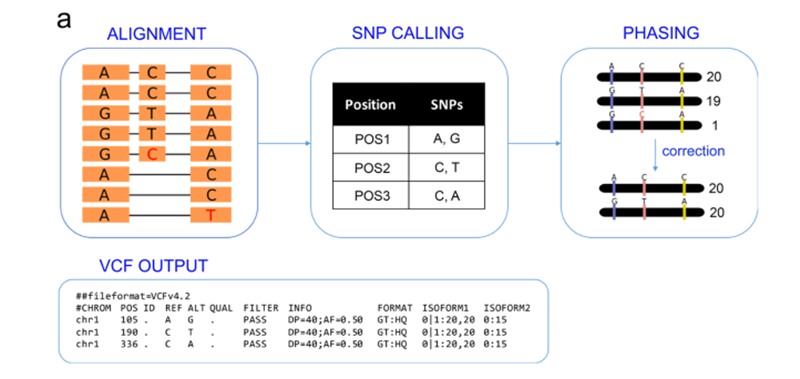
为了对Iso-seq转录本进行分型,作者开发了一套IsoPhase流程,对于每个基因,将全长reads比对到基因区域,然后进行SNP的鉴定。之后使用全长reads的信息重构不同的亚型,使用简单的纠错方案获取两个等位基因。为了分清楚哪个是来自于母本哪个是来至于父本的等位基因,所有的read只能来自于同一个等位基因。只有双亲等位基因被鉴定出来,就得到了F1中每个等位基因的read数。对9000多个基因进行分型后,其中有6700多个基因存在至少一个SNP能够将它归类为B73或者Kill。
使用二代数据进行验证后,其中有96%的SNP得到验证,而其他PacBio特异的SNPs可能二代测序数据对UTR区域的覆盖不够。
等位基因的特异性表达
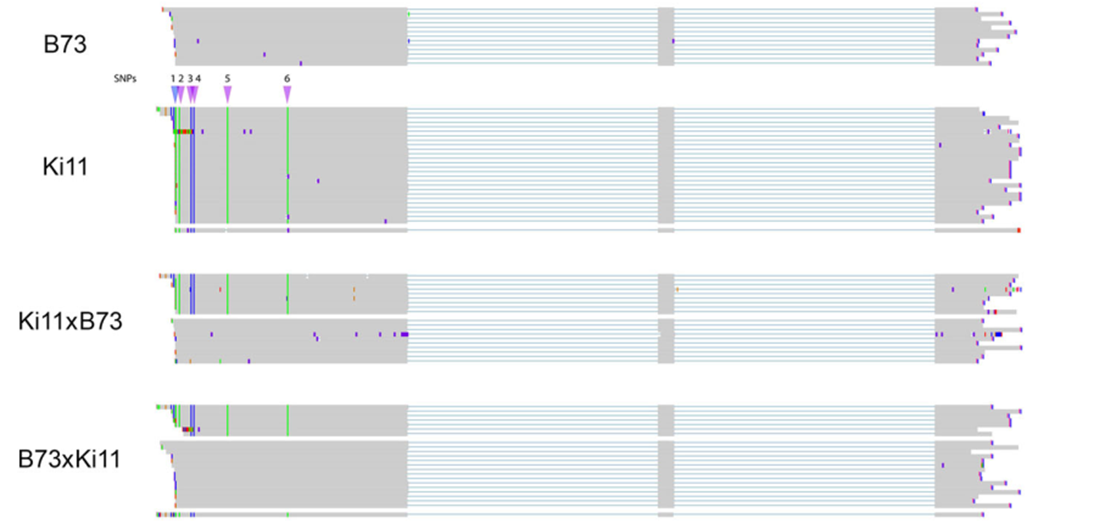
只有母本等位基因的表达,比较了两个双亲在等位基因的表达上的差异
在F1杂交后代中,所有的isform都表达了,但是只有Kill检测出了出了一个isform。
monoallelic genes感觉是纯合的等位基因,在不同组织中比较了这些基因在isform数目上的差异
对SNP进行功能注释
其中有24%的SNP是同义突变,在非同义突变的SNP中, 22,093 多个SNP对 5140 genes 可能存在效应,有将近10~17%是在UTR区域,其中进行容忍度分析发现,0.05值为阀值,大约 2556 genes SNP被认为是有害突变。同时在杂交后代中,isform的数目比父母中更高,但是在胚乳中两个杂交种表现不一致,说明不同的杂交组合影响胚乳的发育。
印记基因、顺式反式调节效应
胚乳中26个父本印记基因,其中70%在之前的研究中被报道了;在胚中2个印记基因同样出现在胚乳中。在胚乳中30个母本印记基因,在根中没有发现印记基因。
顺式、反式调控,通过比较父母本中等位基因表达比率与杂交后代中等位基因的杂交比例来区分。
总结
科学问题非常有趣,研究了不同等位基因特异性的表达,可能是造成杂种优势、印记基因等现象的原因
第一个利用全长转录组数据,在isforms水平对等位基因的特异性表达进行分析
每一个结果都结合了二代测序数据,说明了数据的真实性和可靠性
开发了一套基于全长转录本数据的分析流程IsoPhase
参考
- Variant phasing and haplotypic expression from long-read sequencing in maize

