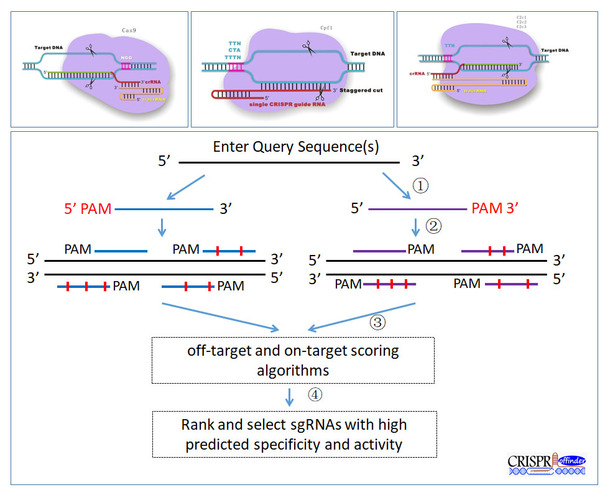
-i <str> Input file -x <int> Length of sgRNA[20] -l <int> The minimum value of GC content [20] -m <int> The maximum value of GC content [80] -g <str> The reference genome sequence -o <str> Searching CRISPR target sites using DNA strands based option(s/a/b) [s, sense strand searching mode] [a, anti-sense strand searching mode] [b, both strand searching mode] -t <str> Type of sgRNA searching mode(s/p) [s, single-gRNA searching mode] [p, paired-gRNA searching mode] -v <str> Operation system(w/l/u/m/a) [w, for windows-32, 64] [l, for linux-64] [u, for linux-32] [m, for MacOSX-64] [a, for MacOSX-32] -n <int> Maximum number of mismatches [5] -s <int> The minimum value of sgRNA offset [-2] 错配罚分 -e <int> The maximum value of sgRNA offset [32] -p <str> Output path
usage: -h|--help print help information -g|--gff= gff file path way -s|--sgRNA= sgRNA file path way -l|--genelength= length of gene -r|--sequence= sgRNA sequence path way -o|--outfile= output file path way