使用教程直接看以下内容
- 客户端软件(软件群文件里有)
- window软件配置
- 安卓配置
- 白名单配置
1.服务端脚本(不需要管):
1 | #修改配置文件 |
重启服务
1 | systemctl restart v2ray |
2.客户端软件
3.windows客户端脚本配置
解压后,开箱即食
使用教程直接看以下内容
- 客户端软件(软件群文件里有)
- window软件配置
- 安卓配置
- 白名单配置
1 | #修改配置文件 |
重启服务
1 | systemctl restart v2ray |
解压后,开箱即食
Plink是一款开源的,用于全基因组关联分析的工具集;它一般用于对基因型数据进行基本的操作。
以下是记录了关联分析中常见的一些操作
--recode将会提取SNP的信息生成对应的map和ped文件--extract则可以进行多个SNP信息的批量提取,后面接一个SNP id的文件
1 | #* 提取单个SNP信息 |
提取指定基因组范围的SNP信息,使用
--extract range 范围文件名;范围文件中包含四列信息分别是
- 染色体编号(可能是数字)
- 起始位置
- 终止位置
- 区域的label(随便起名字)
1 | #* 提取指定范围的SNP信息 |
GEMMA(Genome-wide Efficient Mixed Model Association algorithm) 基于混合线性模型进行全基因组关联分析,相比于其他关联分析软件在以下几点上有所改进:
1. 更快
2. 更加准确
3. 基因plink二进制数据进行分析
4. 功能更加丰富,同时可以进行多个表型的混合线性模型
GEMMA的输入文件可以有两种格式,分别是plink二进制文件和BIMBAM格式;由于plink用的比较普遍,这里只介绍如何使用plink文件格式。
需要注意的是两种格式的文件不能够混用,如果用错了可能会出现未知错误
1 | vcftools --vcf test.vcf --plink --out test |
test.fam文件的第6列代表的是样本对应的表型值,其中每一行代表一个样本;需要注意的是表型文件中样本的顺序要和基因型文件一致.
如果存在多个表型只,可以修改test.fam文件内容,第6列表示第一个表型值,第7列表示第二个表型值;依次类推
Bootstrap(自助法取样)
Bootstrap 的思想是生成一系列 bootstrap 伪样本,每个样本是初始数据有放回抽样。通过对伪样本的计算,获得统计量的分布;当样本数量非常大时,每次抽样中不是重复的样本概率趋近为 0.632,故该抽样方法也叫 0.632 自助法
在正态分布情况下对统计量的置信区间进行估计,在标准正态分布中最常用的就是95%置信区间;从公式上演化推断均值的95%置信区间就是:
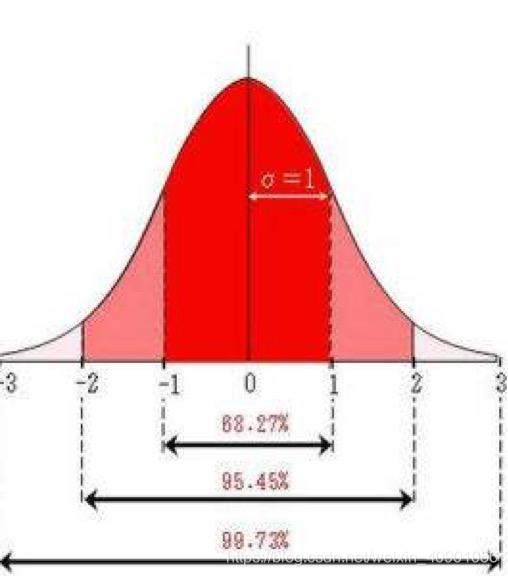
$$\begin{equation}
\mu-1.96\sigma \le X\le\mu+1.96\sigma
\end{equation}$$
但事实上,当找不到合适的分布时,就无法用标准的正态分布计算置信区间了。但幸运的是有一种随机化的方法可以用于计算
非参数分布的置信区间。通过对小样本数据的有放回抽样近似的估计总体的分布,例如我们对包含30个小样本的测试数据进行平均值置信区间估计。
+ 随机进行1000次有放回的抽样,每次抽样从数据集中抽取30个样本
+ 每次抽样后计算当前抽样状态的平均值
+ 最后得到1000次抽样的平均值分布
+ 使用百分位数方法估计平均数的置信区间
1 | #* plink 文件前称 |
1 | #* 提取任意SNP对应的坐标信息 |
神经网络(NN)是在某些输入数据上执行的嵌套函数的集合。 这些函数由参数(由权重和偏差组成)定义,这些参数在 PyTorch 中存储在张量中。
训练 NN 分为两个步骤:
正向传播:在正向传播中,NN 对正确的输出进行最佳猜测。 它通过其每个函数运行输入数据以进行猜测。
反向传播:在反向传播中,NN 根据其猜测中的误差调整其参数。 它通过从输出向后遍历,收集有关函数参数(梯度)的误差导数并使用梯度下降来优化参数来实现。 有关反向传播的更详细的演练,请查看 3Blue1Brown 的视频。
1 | import torch,torchvision |
1 | #* 一个随机张量具有3个通道的64X64的图片 |
1 | prediction=model(data) #forward pass |
1 | loss=(prediction-labels).sum() |
#3.使用SGD优化器根据模型参数的梯度来调整每个模型参数
1 | import torch |
1 | data = [[1, 2], [3, 4]] |
tensor([[1, 2],
[3, 4]])
新的张量可以继承原有张量的结构和数据属性
也可以重新指定新的数据类型
1 | x_ones=torch.ones_like(x_data) #! 保留原张量的结构和属性 |
ones Tensors:
tensor([[1, 1],
[1, 1]])
Random Tensor:
tensor([[0.7318, 0.2529],
[0.1007, 0.8059]])